原标题:原发性噬血细胞综合征涉及基因及部分基因突变定位
来源:转载于 噬血细胞综合征之家 病友论坛 http://
经常会有患者或家属会在群里发出来基因检测报告让大家解读。在这里,我根据王昭主编的《噬血细胞综合征》一书相关知识以及网上相关资料和病友相关实例,结合我自己的理解,聊一下原发性HLH的基因问题,供大家参考。切记切记,更专业的解读还需要找更专业的医生去判断。
噬血细胞综合征(HLH)可大致分为原发性和继发性两种。简单的理解,原发性就是基因有问题,导致免疫系统缺陷,在发挥免疫的过程中,不同的环节出现一些明显的异常问题而引发的噬血现象。属于造娃娃的时候没有造好,先天问题。继发性,是由于后天因为某些因素感染,比如细菌、病毒、寄生虫等而引发的噬血现象。根本原因还是免疫系统能力不足造成的,也有一部分继发的本质还是免疫系统有缺陷,后天的感染也许只是一个引子。这里先把继发的放到一边,聊一下原发的基因问题。
原发性的噬血,一般来讲,分为3类。
家族性HLH
免疫缺陷HLH
EVB驱动HLH。
就我个人理解,这3种都是属于基因造成的免疫缺陷有问题,至于为什么这样分类,我也不清楚。还是我个人理解,不一定正确。家族遗传的,明确上一代有问题或携带致病基因,传给了下一代;免疫缺陷的,上一代没问题,到了下一代这里,以为各种原因基因突变造成了免疫缺陷;在这免疫缺陷中,有一些突变对eb病毒特别敏感,容易引发HLH,便是eb病毒驱动型。
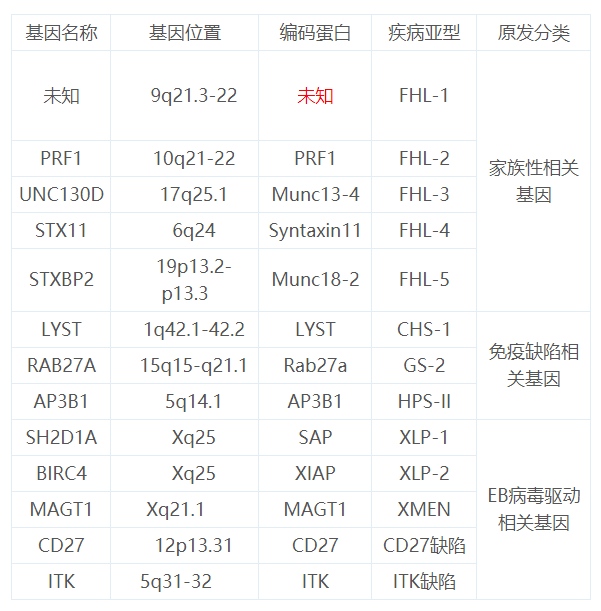
目前来讲,发现和原发HLH的基因有13条,其中家族遗传的第一个亚型FLH-1的基因还不明确外,其余的的12条基因与其编码蛋白以及在染色体的位置如下表。
12条基因与其编码蛋白以及在染色体的位置
所以当我们的基因检查报告出来后,首先要先看看,我们检查的基因包不包括上面的这12条基因。如果有这个12条基因在里面,就好小心的看好了,这些基因的突变位置,以及突变类型,涉不涉及编码蛋白的改变。如果有涉及编码蛋白的改变,基因的可能就比较大了。(如何看基因检查报告,我尽量的学习总结整理一下,以后有机会发出来)我也收集整理了一些这些基因常见的突变的点位,你们可以对照一下。有些检查机构可能只检查和家族遗传相关的,不检查免疫缺陷相关和EB病毒驱动相关的。所以判断是不是基因问题引起的HLH,需要全面检查。
当然,人类大概有不到4万个基因。除了上述明确的12条基因外,可能还有别的没有被发现的基因和HLH有联系,即使检查了这12条基因没问题,也不能完全排除是原发性的可能。
在介绍这些基因之前,先简单介绍一下染色体及其位置的描述。
人类共有22对常染色体和X,Y两条性染色体。一共是46条,其中男人是22对常染色体+XY,女人是22对常染色体+XX。所以生男生女是老爷们来决定的,那些怨老婆生不出儿子的老爷们要检讨了。
之所以叫染色体,是因为最早发现染色体的过程,就是用苏木素,伊红等传统染料对细胞染色,发现有些物质可以被染色,所以叫染色体。染色体是成对出现的,两条染色体连接区域是着丝粒,着丝粒又把染色体分为长短不同的两段,其中长的一段叫长臂,用q表示,短的一段叫短臂,用p表示。每个长臂、短臂细分为区、带,带又细分为亚带次亚带。染色体位置表示就这样来表示,染色体编号1-22或X,Y,长臂或短臂,区号,带号。有亚带或次亚带的,在带号后边加“.”然后写亚带次亚带编号。
举个例子:
FHL-2型噬血的缺陷基因是PRF1,就是我们经常听到的穿孔素基因,它在染色体的位置是10q21-22。依次的含义是
10,表示第10号染色体
Q,表长臂
2表示2区
1表示1带
- 表范围
2 还是表示2区
2 表示1带
10q21-22整体意思就是:PRF1这个基因,在染色体的位置就是10号染色体,长臂上的第2区1带到2区2带这个范围。可以参考XX省XX市XX区XX街道XX号的结构。
闲话少说,具体给大家说说这些基因。(不要崇拜我,下面的基本上属于知识搬家)
首先,是家族噬血。
家族性噬血细胞综合征是常染色体隐性遗传疾病。
共有5个亚型,包括FHL-1、FHL-2、FHL-3、FHL-4和FHL-5,分别存在不同的基因缺陷。
FHL-1:定位于染色体9q21.3-22,但与其相关的潜在缺陷基因及编码蛋白至今仍未被确定。这是第一个被鉴定的FHL基因图谱,在1999年的巴基斯坦的近亲家族中发现。大约占FHL的10%。
(9q21.3中的 .3表示9号染色体长臂2区1带中的第3亚带)
FHL-2:定位于染色体10q21-22,相关缺陷基因为PRF1,编码穿孔素蛋白。约占家族性噬血细胞综合征的20-40%。这个基因又叫穿孔素基因,所编码的蛋白具有打穿细胞壁功能。我想咱这个病,大部分刚刚接触基因检测的听到最多的就是这个穿孔素基因或穿孔素蛋白吧。可以这样理解,有个坏蛋(病毒)跑到了你家(好的细胞)去了,还把窗户门都关上了。我们想抓住这个坏蛋,可能想到的就是在墙壁(细胞壁)上打个孔,然后扔个手雷(溶酶体)进去。这个基因编码的蛋白就是干在墙壁上打孔这件事的。当PRF1基因突变的时候,穿孔素的表达、活性及稳定性下降,受损的穿孔素无法顺利在靶细胞膜上形成管道,造成攻击细胞对靶细胞的杀灭作用受损。目前在PRF1已鉴定了70多个不同的突变位点,最常见的是p.W374X,多与土耳其血统有关。其他常见的如下所列。
PRF1基因常见突变点:c.10C>T(p.R4C);c.272C>T(p.A91V);c.503G>A(p.S168.N);c1122G>A(p.W374X);c.1394C>T(p.T450M);c.50delT(p.L17fsX);c.65delC(p.P22fsX);c.1090-1091delCT(p.T364fsX)。
这些是不是看着眼熟,大部分基因报告里都有这些点位吧,可以参考对照一下。
FHL-3:定位于染色体17q25.1,相关缺陷基因为UNC13D,编码Munc13-4蛋白,约占家族性噬血细胞综合征的17%--30%。Munc13-4的缺陷使得细胞毒颗粒的分泌无法正常启动,穿孔素和颗粒酶不能释放,靶细胞无法被正常杀灭。(能够理解为那个手雷扔不出去)。因为UND13D还影响血小板的颗粒胞吐,所以检测血小板的Munc13-4蛋白表达,有望成为快速筛查FHL-3的新手段。目前已有至少112个突变位点被发现。其常见的如下所列。
UNC130D基因常见突变点:c.1228A>C(p.I410L);c.2588G>A(p.G863D);c.2782C>T(p.R928C);c.2896C>T(p.R966W);c.735+1G>T;c.754-1G>C;c.1055+1G>A;c.1596+1G>C;c.2553+5C>G;c.2346-2349delGGAG(p.R782fsX);c.3229-3235del(p.R1077fsX)
FHL-4:定位于染色体6q24,相关缺陷基因为STX11,编码突触融合蛋白。由于在IL-2刺激下可部分恢复NK细胞脱颗粒和细胞毒性,因此该亚型疾病进程与其他类型FHL相比略为温和。 但其罹患骨髓异常增生综合征及急性白血病的风险较其他类型高。该基因突变多见于库尔德、土耳其、黎巴嫩种族背景的患者中。其常见突变点如下所列。
STX11的常见突变点:c.110delC(p.T37fsX62);c.802C>T(p.Q268X);g.25561-44749;c.369-370delAG;c.374-376delCGC;c.73G>T(p.E25X);c.106G>C(p.E36Q);c.616G>A(p.E206K);c.121C>A(p.L41M);c.326A>G(p.E109G);c.627C>A(p.S209R);c.646C>A(p.R216S);C.799G>A(p.V267M);c.842T>G(p.F281C)
其中,蓝色字体部分多见于中国患者。
FHL-5:定位于染色体19p13.2-p13.3,相关缺陷基因为STXBP2,编码Munc18-2蛋白。约占FHL的5%-20%。纯合子患者1岁以内发病。其在囊泡转运至细胞膜表面的过程中起调节作用,影响NK细胞细胞毒颗粒的胞吐,与STX11存在协同作用。
STXBP2基因的突变点有c.190C>T(p.R64W);c.260delT(p.L87RfsXX32);c.389T>C(p.L130S);c.395G>A(p.R120H);c.395A>C(p.E123A);c.474-473delGA;c.626T>C(p.L209P);c.693-695delGAT(p.I232del);c.706delG(p.A236QfsX24);c.767-771del(p.L257del);c.857G>A(p.R292H);c.953C>T(p.T318>);c.1001C>T(p.P334L);c.1034C>T(pT345M);c.1213C>T(p.R405W);c.1214G>A(p.R405E);c.1247-1G>C(p.V417LfsX126);c.1294C>A(p.Q432X)c.1298C>T(p.A433V);c.1430C>T(p.P477L);c.1621G>A(p.G541S);c.1634C>T(p.S545L);IVS14-1G>C等。
有研究表明,FHL-4FHL-5患者的细胞毒功能能够最终靠IL-2的刺激而部分恢复,目前IL-2已被提出作为这两个亚型的潜在辅助几或用于临时治疗。这对这两个亚型的患者来说,是个福音。
2. 免疫缺陷综合征
Griscelli 综合征2(GS-2):GS是一种常染色体隐性遗传疾病,表现为皮肤和毛发缺失。中文名字叫格里塞利综合征。是由Griscelli在1978年首次报道。可分为3个亚型,GS-1,GS-2,GS-3,其中GS-2与HLH相关。GS-2与定位于染色体15q15-q21.1的RAB27A基因改变有关,表现为局部白化病和神经异常,通常并发致命HLH。RAB27A编码一小段GTP酶,影响细胞毒颗粒缺陷及黑素颗粒的胞吐,并与UNC13D协同,调控造血细胞分泌颗粒。RAB27A突变导致NK细胞和T细胞的严重脱颗粒缺陷,进而导致免疫活性细胞凋亡受阻,同时影响黑素颗粒,神经递质细胞的胞吐及造血细胞颗粒的分泌。检查该类型的HLH除了临床特征之外,通过对毛发的显微镜检查以及判定有核细胞中是不是真的存在巨大颗粒是其诊断根据。
RAB27A突变点有c.11G>T(p.G4V);c.244C>T(p.R82C);c.240-2A>C;c.514del5;c.377delC(p.P126QfsX3);c.109A>T(p.K37X)c.550C>T(p.R184X)。
Chediak-Higashi 综合征(CHS):是常染色体隐性遗传疾病,表现为局部白化病、血小板功能异常、严重免疫缺陷伴HLH。中文名称,柯一里综合征。其基因缺陷为位于染色体1q42.1-q42.2的LYST基因。LYST 蛋白并不参与囊泡融合或分裂,而与囊泡转运的调节有关。由于溶酶体输送障碍导致在黑素细胞和血细胞形成异常巨大颗粒。该病与GS-2表现有所区别,GS的患者毛发呈现较大且不规则的黑素颗粒,主要分布在髓质附近;CHS患者的毛发中,黑素颗粒均匀分布且大于正常毛发中的颗粒。另外,CHS患者的白细胞胞质中存在巨大颗粒,而GS-2患者中没有。
LYST基因常见突变点没有找到。不是没有突变点,是我没有找到相关资料。
Hermansky-Pudlak 综合征2(HPS-2):常染色体隐性遗传疾病,基因缺陷定位于染色体5q14.1,相关缺陷基因为AP3B1,编码结合蛋白复合物3的β3A亚单位。中文名,海一普综合征。表现为眼皮肤型白化病、渐进性肺间质纤维化、出血倾向、反复感染并伴发HLH。HPS-Ⅱ临床表现与CHS-1相似,但无巨大颗粒。
AP3B1基因常见突变点没有找到。不是没有,是我没有找到资料。
3. EBV驱动型HLH
X性联淋巴组织增生综合征(XLP):为X性联遗传性免疫缺陷病,定位于性染色体Xq25。分为两型XLP-1及XLP-2(XIAP),分别对应SH2D1A及BIRC4两种基因突变。以反复发作的HLH、低丙种球蛋白血症和/或淋巴瘤为主要体现。EBV被认为是其发病的驱动因素。SH2D1A编码SAP(信号淋巴细胞激活分子相关蛋白),SAP参与免疫细胞内的信号转导,在淋巴细胞和NK细胞的生长、分化及功能上起直接或间接的及其重要的作用。BIRC4编码XIAP(X性联凋亡抑制蛋白),具有抗凋亡作用,因此,XLP-1患者容易合并淋巴瘤,而XIAP患者罹患淋巴瘤的风险降低。因为是在X染色体上的隐形遗传,所以男性HLH患者要考虑检查SAP和XIAP两种蛋白的表达。如果非发病的女性携带该基因,生出男孩患病的几率为50%。
SH2D1A及BIRC4这两个基因突变点的相关资料我也没找到。
其他类型的EBV驱动型HLH
近年来,一些新的基因缺陷被陆续鉴定,包括IL-2诱导的T细胞激酶缺乏(IL-2–inducible T-cell kinasedeficiency, ITK)、CD27缺乏以及镁离子转运基因(magnesium transporter gene, MAGT1)的突变。这些基因缺陷均以EBV相关的淋巴细胞增殖、淋巴瘤和HLH为主要体现。其中,基因MAGT1位于X染色体上,Xq21.1,也是男性发病的发病患者需要检查的,检查项目为Mg2+转运载体。
还有一些继发性的HLH,也存在着一定的基因背景。这一观点也获得越来越广泛的共识。所以,关于原发继发,除了上述明确的12个基因外,下面一些基因也许考虑。
CCDC141,XIRP2,ARHGAP21,MICAL2,FAM160A2,EXPH5,CADPS2,FKBPL,GD11,KIR2DS5,KIR3DS1,IL10,TGFB,IFNGR1,IFNGR2,NLRC4,IRF5,NLRC4,IL2RG,RAG1,CD127,CD3E,GATA2,CD27,ITK,MAGT1,BTK,FAS,NLRP3,MEFV,TNFRSF1A,IKBKG,STAT1,WAS,ATM,CYBB,22q11.2缺失,LRGUK
上述红色的部分是和免疫缺陷相关的基因。
大家可能看到常染色体隐性遗传疾病标了红色,这涉及到好多病友问的我第一孩子有HLH,能不能生第二的问题。这个等我开新帖聊一下。
本文主要参考《噬血细胞综合征》(王昭主编),以及网上资料和真实噬血案例整理,旨在帮助想对原发HLH了解的患者及家属。不可作为治病判断依据,还是那句话,治病这专业的事情,还得找专业的医护人员来做,我只负责吹牛。
本文里面现有数据如果有医学数据更新或不明不清之处,请大家及时指出修改!
责任编辑:
 万众瞩目:络病大会召开 微血管病变防治开辟新局面
万众瞩目:络病大会召开 微血管病变防治开辟新局面 毓婷x学院奖云课堂完美收官|“毓”青春同行,创意步履不“婷”
毓婷x学院奖云课堂完美收官|“毓”青春同行,创意步履不“婷” 发现就是中晚期的结直肠癌,其实有办法可以避免!
发现就是中晚期的结直肠癌,其实有办法可以避免! “毓”见别YOUNG青春——毓婷携手学院奖2022再启新程
“毓”见别YOUNG青春——毓婷携手学院奖2022再启新程 成都中德肾病医院付冬梅--专业经验技术 让你放心无忧
成都中德肾病医院付冬梅--专业经验技术 让你放心无忧 成都中德肾病医院专家韩艳简介
成都中德肾病医院专家韩艳简介 屈燧林--成都中德肾病医院特聘专家
屈燧林--成都中德肾病医院特聘专家 成都中德肾病医院付冬梅主任:糖尿病肾病应该如何治疗与调养
成都中德肾病医院付冬梅主任:糖尿病肾病应该如何治疗与调养